
Milestone: Identification of prion
Date: April 9, 1982
Location: San Francisco
People: Dr. Stanley Prusiner
On April 9, 1982, doctors at the University of California, San Francisco published a paper in Science showing that an infectious protein causes a neurodegenerative disease in sheep. In doing so, he changed our understanding of how some diseases are transmitted.
“At the time, I was beginning my neurology training, and what struck me most was a disease process in which a patient’s body is not affected by this process and the patient’s brain can be destroyed, resulting in death in two months,” he later said in a speech about his discovery.
you may like
In the 1950s, the term “slow virus” was coined to describe scrapie disease in sheep and goats. By the 1960s, scientists began applying the term to specific human diseases, noting that “kuru,” a disease that struck the Fore tribe of Papua New Guinea, appeared to be transmitted when tribe members ate the brains of people who had previously died from the disease.
Research in chimpanzees in the 1960s showed conclusively that Creutzfeldt-Jakob disease, a fatal and relentless brain disease that appears to run in families, could also be transmitted by feeding chimpanzees with brain tissue from affected animals. When viewed under a microscope, brain tissue affected by kuru, scrapie, and CJD all looked very similar, with a characteristic “spongy” appearance. In other words, the brain tissue became porous, like a sponge.
However, there was a mystery. CJD appeared to run in families. So how can viruses and bacteria be both heritable and infectious?
Prusiner initially studied CJD, but switched his focus to scrapie after seeing data from a team led by radiobiologist Tykva Alper. Alper discovered that scrapie can be transmitted even when infected tissue is exposed to ultraviolet light, which damages DNA.

So Prusiner began studying scrapie in the spleen and brain of mice. However, since symptoms appear within 70 days in hamsters, compared to one to two years in mice, they quickly switched to hamsters. He then worked systematically to isolate and identify the chemical nature of the “infectious agents” that caused the disease.
Eventually, he identified a protein as the culprit.
“Six different lines of evidence indicate that the scrapie pathogen contains proteins necessary for infectivity,” Dr. Prusiner wrote in his seminal 1982 study. All of these showed that disrupting the structure of the protein short-circuits scrapie infection. He went on to show that there was no evidence of the presence of nucleic acids, such as DNA or its analogues, in the samples. He proposed the name “prion” to describe the infectious protein and suggested that it might “encode its own biosynthesis,” adding that “this hypothesis contradicts the ‘central dogma’ of molecular biology.”
What to read next
Prusiner’s proposal was not initially widely accepted. But over the next 15 years, scientists solved the prion’s protein structure and showed that prions can adopt multiple conformations, even when encoded by the same DNA sequence. The researchers also showed how prion shapes resist degradation and that prions can “convert” healthy versions of proteins into diseased forms.
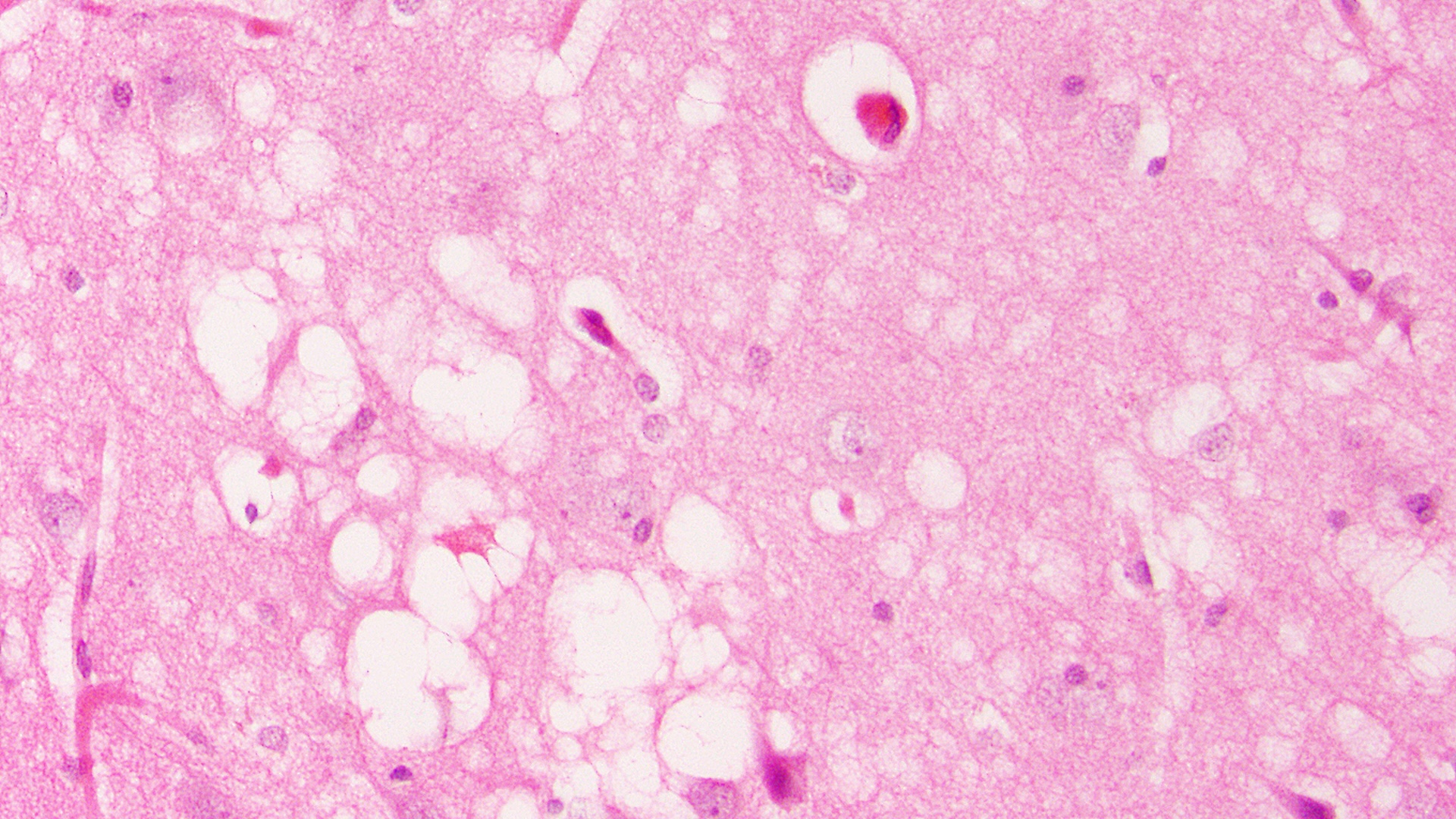
Follow-up studies of familial CJD cases have found that certain genes may predispose people to the disease, and that DNA damage determines the incubation period of the disease.
Prusiner won the Nobel Prize in Physiology or Medicine in 1997 for his research on prions.
Prusiner’s hypothesis was tested in the early 2000s when a mad cow disease epidemic hit the UK. Scientists will ultimately determine that humans became infected after eating beef from cows fed brain tissue from cows infected with bovine spongiform encephalopathy (BSE). Humans who consume meat from cows infected with BSE develop a type of CJD known as “mutant CJD.”
Source link